
کامپیوترهای کوانتومی مجتمع پروتئینی ۱۲۶۳۵ اتمی را شبیهسازی کردند؛ مقیاسی جدید برای کشف دارو
یک گردش کار محاسباتی ترکیبی کوانتومی-کلاسیک با موفقیت بزرگترین سیستم مولکولی با اهمیت بیولوژیکی را تا به امروز مدلسازی کرده است که نشاندهنده افزایش ۴۰ برابری در مقیاس شبیهسازی است. این پیشرفت نشان میدهد که چگونه پردازندههای کوانتومی میتوانند تعاملات پیچیده الکترونی را که در قلب تحقیقات دارویی قرار دارند، مدیریت کنند.
به قلم ساناز امامی
این خبر را به اشتراک بگذارید

- توسعهدهندگان سختافزار کوانتومی
- تمرکز بر مقیاسدهی سریع ابرمحاسبات کوانتوممحور و ادغام موفقیتآمیز پردازندههای کوانتومی (QPU) با مراکز داده کلاسیک.
- زیستشناسان محاسباتی
- تأکید بر پیشرفتهای الگوریتمی که امکان به کارگیری روشهای کوانتومی را برای مجتمعهای پروتئینی مرتبط با زیستشناسی و محلول در آب فراهم میکند.
- تحلیلگران مراقبتهای بهداشتی
- حفظ دیدگاهی محتاطانه، با اشاره به اینکه اگرچه مقیاس محاسباتی چشمگیر است، اما این فناوری هنوز در مرحله پیشبالینی است و هنوز از روشهای کلاسیک پیشی نگرفته است.
نکات کلیدی
- یک تیم تحقیقاتی بینالمللی با استفاده از رویکرد محاسباتی ترکیبی کوانتومی-کلاسیک، یک مجتمع پروتئین-لیگاند ۱۲۶۳۵ اتمی را شبیهسازی کرده است.
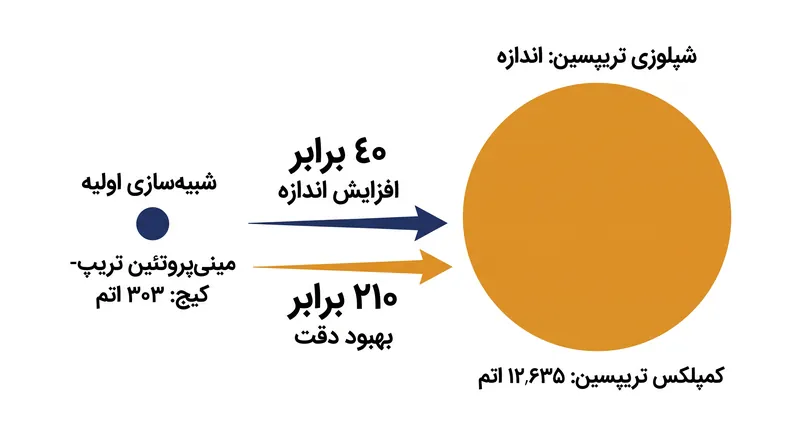
- این دستاورد نشاندهنده افزایش ۴۰ برابری در اندازه سیستم و بهبود ۲۱۰ برابری در دقت نسبت به معیارهایی است که تنها چهار ماه قبل تعیین شده بودند.
- ابرکامپیوترهای کلاسیک مولکول را به قطعات تقسیم کردند، در حالی که پردازندههای کوانتومی IBM پیچیدهترین مناطق بسیار درهمتنیده را محاسبه کردند.
- این شبیهسازی تریپسین و T4-لیزوزیم را در محلول آب مایع مدلسازی کرد و محیطهای بیولوژیکی طبیعی آنها را تقلید نمود.
- اگرچه هنوز سریعتر از روشهای کاملاً کلاسیک نیست، اما مسیر سریع پیشرفت نشان میدهد که محاسبات کوانتومی به زودی کشف داروهای دارویی را تسریع خواهد کرد.
چرا مهم است
پیشبینی دقیق نحوه اتصال یک داروی کاندید به پروتئین هدف، در حال حاضر سالها آزمون و خطا میطلبد. محققان با اثبات اینکه سختافزار کوانتومی میتواند پیچیدگی عظیم این تعاملات مولکولی را مدیریت کند، در حال ایجاد زیرساختی هستند تا مسیر توسعه دارویی که یک دهه طول میکشد را به شدت کوتاه کنند.
روند رویداد
اوایل ۲۰۲۶
محققان مینیپروتئین ۳۰۳ اتمی Trp-cage را شبیهسازی میکنند و خط مبنایی برای ابرمحاسبات کوانتوممحور ایجاد میکنند.
مه ۲۰۲۶
همکاری کلینیک کلیولند، ریکِن و آیبیام با موفقیت مجتمع تریپسین ۱۲۶۳۵ اتمی را مدلسازی میکند.
جولای ۲۰۲۶
جامعه علمی پتانسیل الگوریتم EWF-TrimSQD را برای دور زدن تنگناهای محاسباتی سنتی در کشف دارو تحلیل میکند.
محققان کلینیک کلیولند، ریکِن و آیبیام با استفاده از یک رویکرد محاسباتی ترکیبی کوانتومی-کلاسیک، ساختار الکترونیکی یک مجتمع پروتئین-لیگاند حاوی ۱۲۶۳۵ اتم را با موفقیت شبیهسازی کردند.
مولکولهای خاصی که مدلسازی شدند، تریپسین (یک آنزیم گوارشی) متصل به یک مهارکننده، و T4-لیزوزیم (یک پروتئین سیستم ایمنی) بودند. هر دو در محلول آب مایع شبیهسازی شدند تا محیط بیولوژیکی طبیعی خود را تقلید کنند.[1]
این دستاورد بزرگترین شبیهسازی مولکولی با اهمیت بیولوژیکی است که تاکنون بر روی سختافزار کوانتومی انجام شده و رکورد قبلی یک مینیپروتئین ۳۰۳ اتمی را که تنها چهار ماه پیش ثبت شده بود، شکست.
چالش اصلی در کشف محاسباتی دارو، پیشبینی دقیق نحوه اتصال یک مولکول کوچک، معروف به لیگاند یا داروی کاندید، به یک پروتئین هدف بزرگ است.[3]
کامپیوترهای کلاسیک با این کار مشکل دارند، زیرا «همبستگی الکترونی»—تعاملات مکانیک کوانتومی پیچیده و درهمتنیده بین الکترونها که نحوه اتصال و واکنش مولکولها را تعیین میکند—محاسبه را دشوار میسازد.
برای غلبه بر این تنگنای محاسباتی، تیم بینالمللی از چارچوبی به نام ابرمحاسبات کوانتوممحور (QCSC) استفاده کرد که بخشهای مختلف شبیهسازی را به ماشینهایی که برای آن کار مناسبتر هستند، واگذار میکند.[2]
این گردش کار متکی بر یک رویکرد الگوریتمی جدید به نام EWF-TrimSQD (تابع موج جاسازی شده – قطریسازی کوانتومی مبتنی بر نمونهبرداری پیرایش شده) است.[2]
ابرکامپیوترهای کلاسیک—به طور خاص فوگاکو (Fugaku) متعلق به ریکِن و میابی-جی (Miyabi-G) متعلق به دانشگاه توکیو—ابتدا مجتمع عظیم پروتئین-لیگاند را به قطعات کوچکتر و قابل محاسبه تجزیه میکنند.[2]
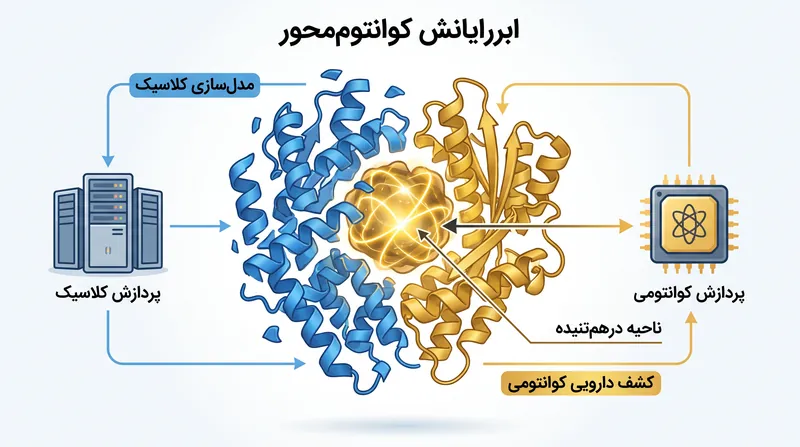
سیستمهای کلاسیک مناطق سادهتر و کمتر درهمتنیده مولکول را مدیریت میکنند. پیچیدهترین خوشهها، جایی که همبستگی الکترونی قویتر است، به پردازندههای کوانتومی منتقل میشوند.[2]
سیستمهای کلاسیک مناطق سادهتر و کمتر درهمتنیده مولکول را مدیریت میکنند.
دو پردازنده ۱۵۶ کیوبیتی IBM Quantum Heron r2 نمونهبرداری کوانتومی را انجام دادند. این پردازندهها با استفاده از حداکثر ۹۴ کیوبیت، ۹۲۰۰ مدار را طی ۱۰۰ ساعت اجرا کردند و ۱.۳ میلیارد نتیجه اندازهگیری را جمعآوری نمودند.
یک پیشرفت الگوریتمی حیاتی به تیم اجازه داد تا شبیهسازی را بدون تحمیل هزینههای محاسباتی تصاعدی، مقیاسدهی کند. محققان دریافتند که همبستگیهای مکانیک کوانتومی فراتر از یک حوزه محلی ۷ تا ۱۰ آنگستروم ناچیز میشوند.[2]
با محدود کردن محاسبات کوانتومی به این حوزههای محلی، تیم به افزایش ۴۰ برابری در اندازه سیستم و بهبود ۲۱۰ برابری در دقت برای یک مرحله کلیدی گردش کار در مقایسه با روشهای قبلی دست یافت.

دکتر کنت مرز، نویسنده اصلی این مطالعه از کلینیک کلیولند، اشاره کرد که عبور از مانع ۱۲۰۰۰ اتمی، چارچوبی عملی برای به کارگیری روشهای کوانتومی در مسائل بیولوژیکی مرتبط با علم را نشان میدهد.[1][3]
صنعت داروسازی این پیشرفتها را از نزدیک زیر نظر دارد، زیرا محاسبه دقیق انرژیها و حرکات مولکولی در مراحل اولیه کشف میتواند به شدت جدول زمانی توسعه دارو را که در حال حاضر بیش از یک دهه طول میکشد، کوتاه کند.[3]
با این حال، محققان و تحلیلگران بر عدم قطعیت شفاف در مورد کاربردهای بالینی فوری تأکید میکنند. روش ترکیبی فعلی هنوز از نظر سرعت یا کارایی هزینه، بهتر از بهترین رویکردهای کاملاً کلاسیک برای شیمی پروتئین عمل نمیکند.
علاوه بر این، شبیهسازی موفقیتآمیز یک مجتمع پروتئین-لیگاند یک نقطه عطف محاسباتی است، نه یک نتیجه تأیید شده در کشف دارو. کاربردهای مراقبتهای بهداشتی محاسبات کوانتومی همچنان به شدت در مراحل پیشبالینی و محاسباتی باقی میماند.
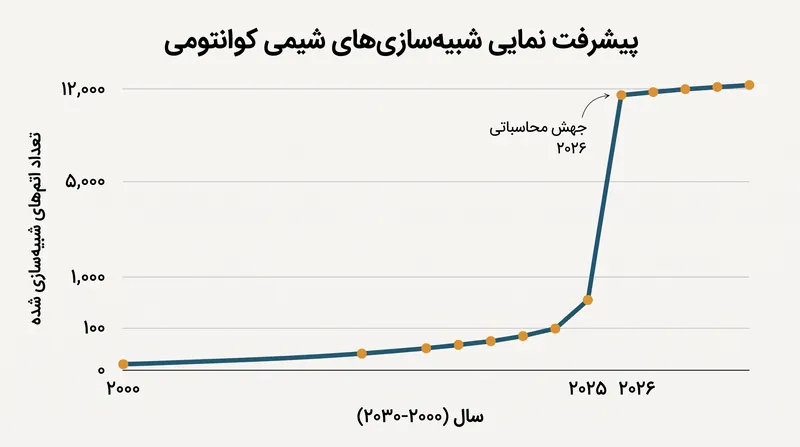
با وجود این هشدارها، مسیر بهبود قابل توجه است. حرکت از یک سیستم ۳۰۳ اتمی در خلاء به یک سیستم ۱۲۶۳۵ اتمی در محلول آبی طی چند ماه نشان میدهد که رویکردهای کوانتوممحور میتوانند در آینده نزدیک با جایگزینهای کلاسیک رقابتی شوند.
با ادامه تکامل سختافزار کوانتومی مقاوم در برابر خطا، انتظار میرود این گردش کار ترکیبی بیشتر مقیاسپذیر شود و به طور بالقوه مدلسازی پیشبینیکننده کل سیستمهای بیولوژیکی را ممکن سازد و نحوه طراحی داروها را به طور اساسی تغییر دهد.
بررسی عمیق دیدگاهها
شیمیدانان محاسباتی
تمرکز بر جهش الگوریتمی که تنگناهای سنتی شبیهسازی مولکولی را دور زد.
برای شیمیدانان محاسباتی، سختافزار در درجه دوم اهمیت قرار دارد و پیشرفت الگوریتمی EWF-TrimSQD مهمتر است. محققان با درک اینکه همبستگیهای مکانیک کوانتومی فراتر از شعاع ۷ تا ۱۰ آنگستروم ناچیز میشوند، توانستند بار کاری پردازنده کوانتومی را به یک حوزه محلی محدود کنند. این روش مقیاسدهی خطی، تنگنای «مقیاسدهی توان پنجم» را که به طور سنتی شبیهسازی مولکولهای بزرگ را به صورت تصاعدی پرهزینهتر میکرد، دور زد و ثابت کرد که الگوریتمهای ترکیبی میتوانند بر محدودیتهای سختافزاری خام غلبه کنند.
صنعت داروسازی
تمرکز بر وعده بلندمدت مدلسازی پیشبینیکننده برای کاهش مسیر یک دههای کشف دارو.
بخش داروسازی این نقطه عطف را از دریچه زمان و سرمایه میبیند. در حال حاضر، شناسایی نحوه اتصال یک داروی کاندید به پروتئین هدف، سالها آزمون و خطا میطلبد و به هزینه میلیاردی عرضه یک داروی جدید به بازار کمک میکند. اگر ابرمحاسبات کوانتوممحور بتواند این انرژیهای اتصال را به طور دقیق به صورت مجازی (in silico) پیشبینی کند، شرکتهای داروسازی میتوانند میلیونها ترکیب را به صورت مجازی غربالگری کنند و زمان صرف شده در آزمایشهای آزمایشگاهی پیشبالینی را به شدت کاهش دهند.
واقعگرایان کوانتومی
تمرکز بر محدودیتهای فعلی، با اشاره به اینکه این سیستم هنوز از نظر سرعت یا هزینه از ابرکامپیوترهای کلاسیک پیشی نگرفته است.
تحلیلگران و شکاکان کوانتومی تأکید میکنند که اگرچه شبیهسازی ۱۲۶۳۵ اتمی یک اثبات مفهوم تاریخی است، اما هنوز جایگزین عملی برای ابزارهای موجود نیست. گردش کار ترکیبی به بیش از ۱۰۰ ساعت نمونهبرداری کوانتومی و منابع عظیم ابرکامپیوتری نیاز داشت و هنوز هم از بهترین روشهای کاملاً کلاسیک برای شیمی پروتئین بهتر عمل نمیکند. آنها هشدار میدهند که کاربرد بالینی واقعی و «مزیت کوانتومی» در کشف دارو، تا زمان ورود سختافزار کوانتومی کاملاً مقاوم در برابر خطا، سالها فاصله دارد.
اصطلاحات کلیدی
- ابرمحاسبات کوانتوممحور (QCSC)
- یک گردش کار ترکیبی که ابرکامپیوترهای کلاسیک را با پردازندههای کوانتومی ادغام میکند تا مسائلی را حل کند که هیچکدام به تنهایی قادر به حل آنها نیستند.
- لیگاند
- یک مولکول کوچک، مانند یک داروی کاندید، که به یک محل خاص روی پروتئین هدف متصل میشود تا یک پاسخ بیولوژیکی را آغاز یا مسدود کند.
- همبستگی الکترونی
- حرکت و تعامل پیچیده و وابسته به هم الکترونها در یک مولکول، که نحوه اتصال و واکنش مولکولها را تعیین میکند.
- کیوبیت
- واحد اصلی اطلاعات کوانتومی، که برخلاف بیتهای کلاسیک که فقط ۰ یا ۱ هستند، قادر به نمایش همزمان چندین حالت است.
- آنگستروم
- واحدی برای طول برابر با یک دهمیلیاردم متر، که معمولاً برای اندازهگیری اتمها و ساختارهای مولکولی استفاده میشود.
آنچه نمیدانیم
- اینکه چه زمانی ابرمحاسبات کوانتوممحور به طور قطعی از بهترین روشهای کاملاً کلاسیک از نظر سرعت و هزینه در شیمی پروتئین پیشی خواهد گرفت.
- این گردش کار ترکیبی با چه سهولتی به سیستمهای بیولوژیکی بزرگتر، مانند کل مسیرهای سلولی، مقیاسپذیر خواهد بود.
- آیا صنعت داروسازی این گردشهای کار کوانتومی را برای غربالگری اولیه دارو پیش از رسیدن کامپیوترهای کوانتومی مقاوم در برابر خطا به کار خواهد گرفت یا خیر.
پرسشهای متداول
مجتمع پروتئین-لیگاند چیست؟
ساختاری است که وقتی یک مولکول کوچکتر (لیگاند، که اغلب یک داروی کاندید است) به یک محل خاص روی یک پروتئین هدف بزرگتر متصل میشود، تشکیل شده و رفتار بیولوژیکی آن را تغییر میدهد.
چرا کامپیوترهای کلاسیک با این کار مشکل دارند؟
با بزرگ شدن مولکولها، تعاملات مکانیک کوانتومی بین الکترونهای آنها—معروف به همبستگی الکترونی—به طور تصاعدی پیچیدهتر میشوند و محاسبه دقیق آنها پردازندههای کلاسیک را تحت فشار قرار میدهد.
آیا این کامپیوتر کوانتومی در حال حاضر داروهای جدید طراحی میکند؟
خیر. این یک نقطه عطف محاسباتی پیشبالینی است که نشان میدهد سختافزار و الگوریتمها میتوانند مقیاس مولکولهای بیولوژیکی را مدیریت کنند، اما هنوز داروهای تأیید شده تولید نمیکند.
ابرمحاسبات کوانتوممحور چیست؟
یک رویکرد ترکیبی است که در آن ابرکامپیوترهای کلاسیک بخش عمدهای از یک مسئله را مدیریت میکنند و تنها پیچیدهترین محاسبات بسیار درهمتنیده را به یک پردازنده کوانتومی واگذار میکنند.
منابع
پوشش منابع
3 منبع
3 دیدگاه شناساییشده


[1]Chemistry Worldزیستشناسان محاسباتی
Two IBM quantum processors working in concert with two supercomputers simulate a protein–ligand system with 12000 atoms
مطالعه در Chemistry World →[2]Quantum Computing Reportزیستشناسان محاسباتی
Cleveland Clinic, RIKEN, and IBM Simulate 12,635-Atom Protein Complex
مطالعه در Quantum Computing Report →[3]WKYCزیستشناسان محاسباتی
Cleveland Clinic just did something that's never been done before
مطالعه در WKYC →
نظرات



هر زاویه. هر روز.
دریافت علم اخبار همراه با پوشش کامل منابع و تحلیل دیدگاهها، مستقیم در صندوق ورودی شما.